Research
Developing novel computational and statistical methods for genetic studies in large biobanks
With the emergence of large-scale biobanks comes the opportunity to perform phenome-wide scans (PheWAS) of genetic associations for complex human diseases and traits, that is, to carry out a GWAS on all available phenotypes. In practice, this translates to testing associations between tens of millions of genetic variants and thousands of phenotypes, which pose computational and analytical challenges. To address these challenges, we have developed widely used statistical tools to enable the studies in large-scale biobanks and cohorts to uncover common and rare genetic variants that are associated with human disease susceptibility and progression. These include SAIGE for GWAS on binary phenotypes for disease risk, GATE for GWAS on time-to-event phenotypes for disease progression, as well as SAIGE-GENE and SAIGE-GENE+ for rare variant association studies.
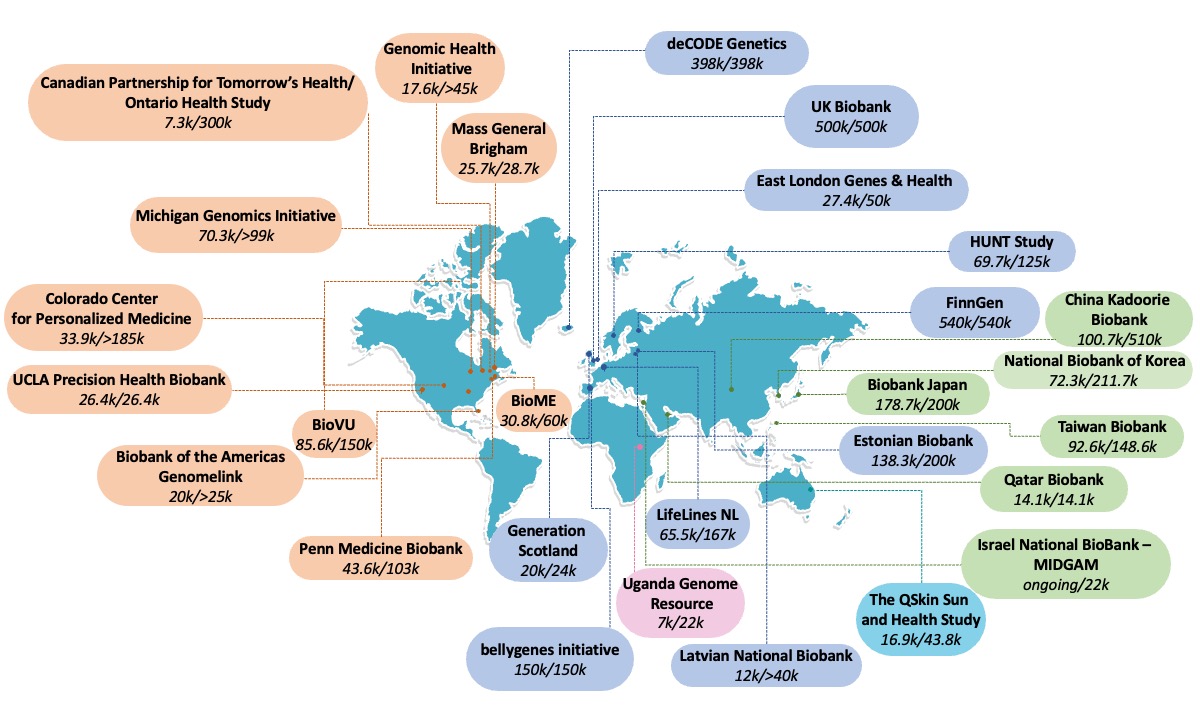
Understanding the genetic etiology of human diseases and traits by meta-analyzing large biobanks
Biobanks with health data linked with genomic information provide resources for the genetic research community. The drop in the cost of genotyping and sequencing has led to an increase in the number of genomically profiled biobanks worldwide. Compared with disease- or trait-based cohorts centered around a particular phenotype or several relevant phenotypes, biobanks enable cost-effective genetic discovery for hundreds to thousands of phenotypes, curated from electronic health records (EHRs), registry-based data (e.g., pharmaceutical, death, or cancer registry data), and/or epidemiological questionnaires to understand the genetic etiology of human diseases. We apply the cutting-edge statistical tools for identifying novel genetic risk factors for human diseases in large biobanks, including the FinnGen study, HUNT, UK Biobank and All of US. We are co-leading the Global Biobank Meta-analysis Initiative (GBMI) — a collaborative network of more than 20 biobanks from 5 continents representing more than 2.6 million consented individuals with genetic data linked to electronic health records. We also actively help coordinate the analyses of biobank meta-analyses for rare genetic variants in the BraVa consortium.
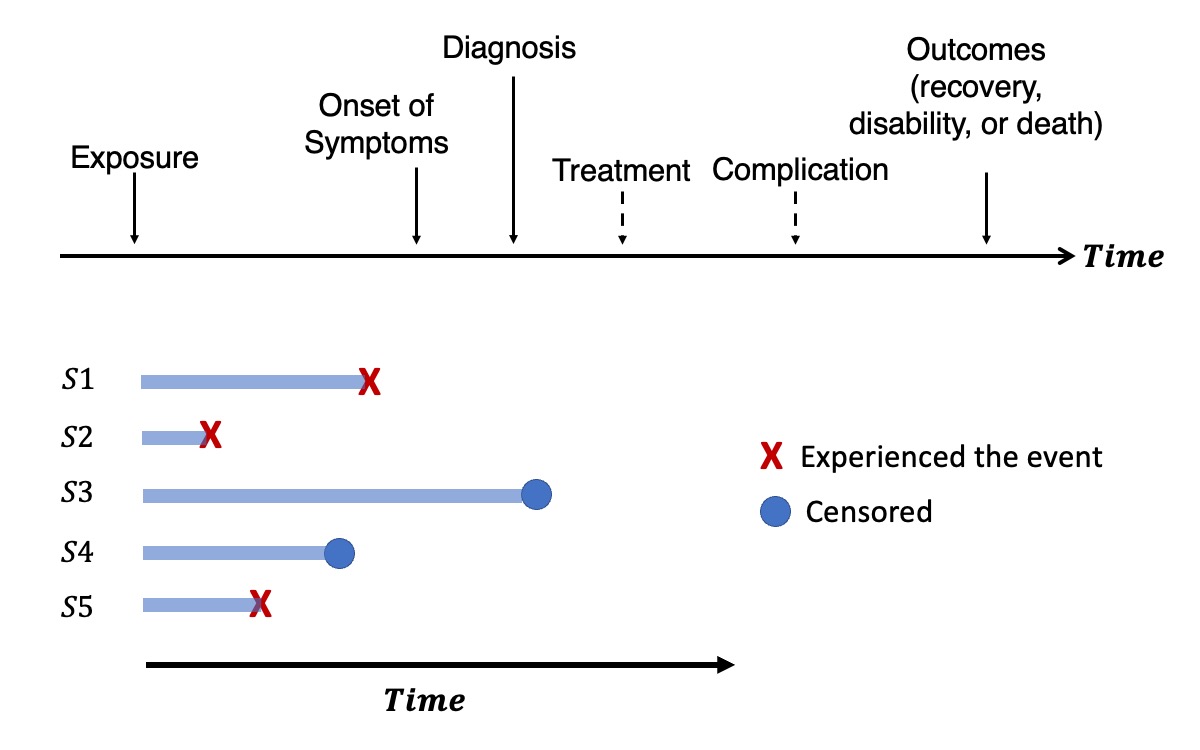
Understanding the genetic etiology of human diseases onset and progression
Genome-wide survival association studies of censored time-to-event (TTE) phenotypes can identify genetic variants associated with lifespan and disease progression. Large biobanks, such as UK Biobank and FinnGen, with decades of EHRs linked to genomic data, provide opportunities to such studies for a comprehensive understanding of genetic basis of disease course over time. However, analytical challenges exist for such studies, such as high computation cost to handle large data sizes with sample relatedness and heavy data censoring. We are interested in deveoping statistical and computational methods to facilitate the genetic studies of diseases onset and progression as well as applying these tools to analyze large biobanks.
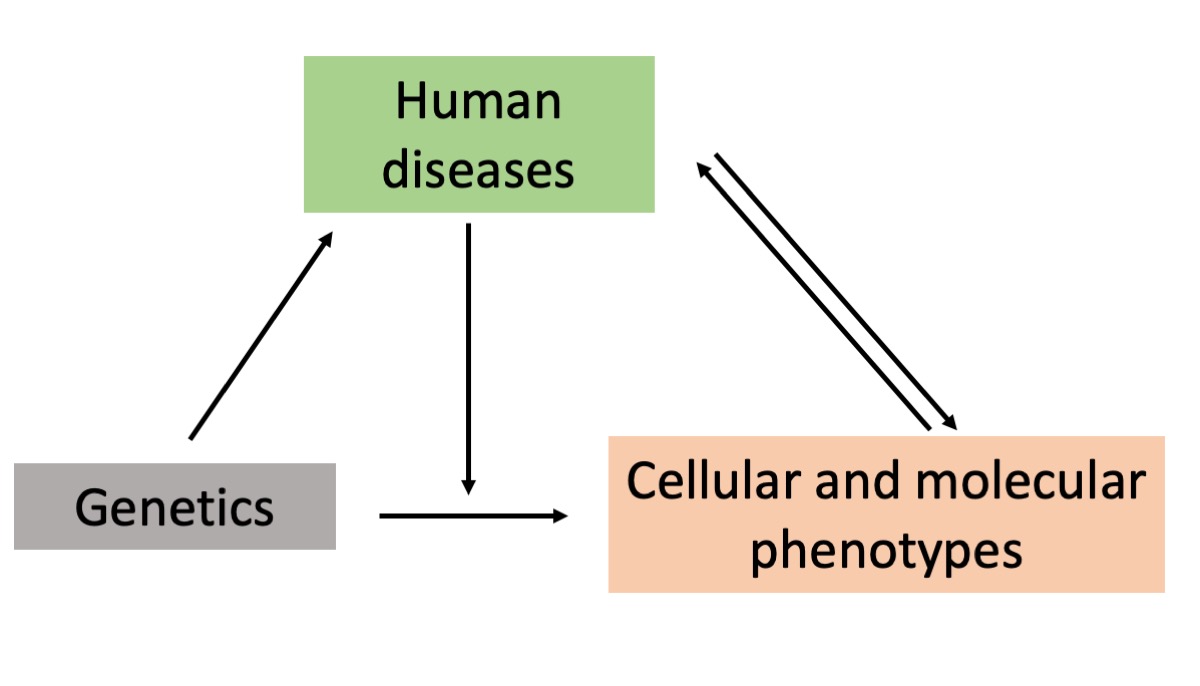
Interpreting the genetic association discoveries for functional follow-up by leveraging multi-omics data
Hundreds of thousands of disease-associated genomic variations have been uncovered within the past two decades, but for most of these discoveries, the molecular and functional consequences remain unknown, representing a fundamental challenge for the translation of these discoveries into biological insight and medical value. We are interested in bridging the gap from complex trait association to relevant biological processes through systematic genetic analysis of high-dimensional molecular and cellular datasets such as transcriptomics, epigenomics, proteomics, and metabolomics, and combining multi-omics data to understand the molecular mechanisms underlying human diseases. Currently, we are focusing on developing and applying scalable and efficient statistical methods for eQTL mapping at the single-cell level.
People

Wei Zhou
wzhou@broadinstitute.org Principal Investigator
Wei is an Assistant Professor in the Center for Genomic Medicine at Massachusetts General Hospital
and Associate member in the Stanley Center for Psychiatric Research at Broad Institute of MIT and Harvard. She is co-leading the Global Biobank Meta-analysis Initiative (GBMI) and is on the Organizing Committee of
International Common Disease Alliance (ICDA).
As a statistical geneticist, Wei's research has mainly focused on developing and applying statistical methods to investigate the underlying genetic basis of complex human diseases and traits by leveraging large-scale biobanks and data sets.

Grant Chau
gchau@broadinstitute.org Associate Computational Biologist
Grant is an Associate Computational Biologist I in Dr. Benjamin Neale's Lab. He is working with Wei on the project to develop a novel efficient method to mapping eQTLs at the single-cell resolution. He is interested in leveraging numerical and statistical methods to estimate genetic contributions to psychological traits. He graduated from Northeastern University in 2022 with a BS in Mathematics and Computer Science.

Ying Wang
yiwang@broadinstitute.org Postdoctoral Fellow
Ying is a postdoctoral research fellow in Dr. Alicia Martin’s lab. She is working with Wei to develop a novel statistial method to map rare genetic variations for time-to-event phenotypes. She did her PhD in statistical/quantitative genetics at the University of Queensland. During her PhD, she worked on the application of genomic prediction methods in diverse ancestry populations and development of such methods to improve transferability of polygenic scores in trans-ancestry predictions. She has a broad interest in research areas related to multi-ancestry studies and population genetics.
Publications
Selected publications are below. For a full list of publications, please see Google scholar.
- Zhou, W.,Kanai, M., Wu, K.-H. H., Rasheed, H., Tsuo, K., Hirbo, J. B., Wang, Y., Bhattacharya, A., Zhao, H., Namba, S., Surakka, I., Wolford, B. N., Lo Faro, V., Lopera-Maya, E. A., Läll, K., Favé, M.-J., Partanen, J. J., Chapman, S. B., Karjalainen, J., … Neale, B. M. (2022) Global Biobank Meta-analysis Initiative: Powering genetic discovery across human disease. Cell Genomics.
- Zhou, W*, Bi W*, Zhao Z*, Dey KK, Jagadeesh KA, Karczewski KJ, Daly MJ, Neale BM, Lee S.(2022) SAIGE-GENE+ improves the efficiency and accuracy of set-based rare variant association tests. Nat Genet. 54(10):1466-1469. PMID: 36138231. PMCID: PMC9534766
- Kurki, M. I., Karjalainen, J., Palta, P., Sipilä, T. P., Kristiansson, K., Donner, K. M., ...,Zhou, W, ... & Waring, J. (2023). FinnGen provides genetic insights from a well-phenotyped isolated population. Nature, 613(7944), 508-518.
- Dey, R.*, Zhou, W.*, Kiiskinen, T., Havulinna, A., Elliott, A., Karjalainen, J., ... & Lin, X. (2022). Efficient and accurate frailty model approach for genome-wide survival association analysis in large-scale biobanks. Nature communications, 13(1), 5437.
- Zhou, W.*, Brumpton, B.*, Kabil, O.*, Gudmundsson, J.*, Thorleifsson, G.*, Weinstock, J., ... & Åsvold, B. O. (2020). GWAS of thyroid stimulating hormone highlights pleiotropic effects and inverse association with thyroid cancer. Nature communications, 11(1), 3981.
- Zhou, W.*, Zhao, Z.*, Nielsen, J. B., Fritsche, L. G., LeFaive, J., Gagliano Taliun, S. A., ... & Lee, S. (2020). Scalable generalized linear mixed model for region-based association tests in large biobanks and cohorts. Nature genetics, 52(6), 634-639.
- Zhou, W., Nielsen, J. B., Fritsche, L. G., Dey, R., Gabrielsen, M. E., Wolford, B. N., ... & Lee, S. (2018). Efficiently controlling for case-control imbalance and sample relatedness in large-scale genetic association studies. Nature genetics, 50(9), 1335-1341.
Resources
SAIGE
SAIGE is an R-package for GWAS with adjusting for sample relatedness and case-control imbalance. It can analyze very large sample data (ex. ~400,000 samples in UKBiobank) and produce accurate p-values by using saddlepoint approximation.
Paper here.
Tutorial here.
Workshop video here.
Github repo here.
Browse summary statistics of GWAS for 1403 ICD-based traits using SAIGE in the UK Biobank here.
SAIGE-GENE(+)
SAIGE-GENE (now known as SAIGE-GENE+) are new method extension in the R package for testing rare variant in set-based tests in large biobanks and cohorts.
Paper here. and here.
Tutorial here.
Workshop video here.
Github repo here.
Browse summary statistics of gene-based association studies for >4,000 traits using SAIGE-GENE in the UK Biobank here.
GATE
GATE (Genetic Analysis of Time-to-Event phenotypes) is an R package with Scalable and accurate genome-wide association analysis of censored survival data in large scale biobanks using frailty models.GATE performs single-variant association tests for time-to-event endpoints and uses the saddlepoint approximation(SPA)(mhof, J. P. , 1961; Kuonen, D. 1999; Dey, R. et.al 2017) to account for heavy censoring rates.
Paper here.
Github repo here.

Join us
We are inviting applications for two postdoctoral fellows.
One position emphasizes leading the projects in the Global Biobank Meta-analysis Initiative (GBMI) to study the genetics of complex disease progression over time by integrating longitudinal phenotypes across more than 20 biobanks, comprising over 2.4 million individuals. Since its initiation in 2019, GBMI has rapidly evolved into a highly active collaboration network and published over 14 high-impact journal papers from its first round of projects. Collaborations with global researchers and biobanks offer exceptional opportunities for research, training, and networking. The successful applicant will closely work with Dr. Wei Zhou, Dr. Ida Surakka, and the GBMI coordination team.
The other position emphasizes developing and applying cutting-edge scalable and efficient computational and statistical tools for uncovering genetic and genomic risk factors of complex human diseases using large-scale high-dimensional genetics and omics data. Specific areas of interest include, but are not limited to, 1) understand genetics for human disease onset and progression, 2) map and harmonize genetics of rare and common variations for molecular phenotypes measured at single-cell and/or bulk resolution, 3) prioritize functional variants and genes by integrating epigenetics and xQTLs to GWAS.
Environment/Lab Culture:
The Zhou Group is a new lab in the Center for Genomic Medicine at the Massachusetts General Hospital and the Stanley Center for Psychiatric Research at Broad Institute of MIT and Harvard. Our community consists of an interdisciplinary group of postdoctoral fellows, scientists, bioinformaticians, computational biologists, and graduate students who work together in a mutually supportive and respectful environment. Postdoctoral fellows in these positions will be embedded in this small, new lab, benefiting from highly personalized mentoring, but also have access to the range of expertise within the centers, including but not limited to Ben Neale, Mark Daly, Alicia Martin, Konrad Karczewski, Tian Ge, Hailiang Huang and their respective groups. For those interested, joint mentorship can be pursued from both Dr. Zhou and other faculty members. We are also an engaged member of the larger Broad community, and we maintain close ties with scientific communities at MIT- and Harvard-based institutions, and collaborators across the world.
Postdocs will have the opportunity to play an active role in shaping the lab culture, where ideas are freely shared, and contributions are highly valued. Dr. Zhou places a high priority on mentoring trainees to work toward achieving their career paths and goals. Each team member is encouraged to identify and adopt their most effective work style, while maintaining respect for the preferences of others
Requirements:
PhD in biostatistics, bioinformatics, computational biology, computer science, human genetics, statistics, mathematics, or related quantitative discipline. The candidate should have a strong computational background, demonstrated proficiency in Python, R, or other programming languages, experience in working in HPC and/or cloud-based environment and experience in genetic/genomics oriented research.
How to apply:
Interested applicants please send a CV, a short research statement, and names of three referees to
wzhou@broadinstitute.org